
Latest Research
- 2014.12.26
Revealing "Rendezvous of proteins" by the engineering of firefly glowing mechanism
~Development of a novel protein-protein interaction assay~
Introduction
It is almost 10 years since the human genome sequence was clarified. However, the mystery of life is not yet elucidated, and the causes of various diseases remain unknown. One of the reason is , the insufficient understanding of protein functions, in particular, the interaction between proteins. Currently, in human cells, 150-300 thousand kinds of protein-protein interactions (PPIs) exist. Understanding of PPIs and their temporal and spatial changes and the control mechanism are not only essential in biology, but are also extremely important for understanding and preventing diseases. The importance of PPI assay is well recognized by a number of predecessors, and a number of assay/selection systems have been devised. For example, Protein-fragment Complementation Assay (PCA) is a well-known method primarily used in the cell. In PCA, probe proteins are made by dividing the reporter protein such as an enzyme or fluorescent protein into two and fused respectively to the interaction partner. These probes are in close proximity at the time of interaction, and refolding of the reporter protein and subsequent recovery of its activity is used to detect interaction. It shows high signal / background (S/B) ratio, and has been used in the cell. However, detecting interaction of purified probes in vitro by PCA was rather difficult, due to the instability of split protein probes. As an alternative PPI assay, we recently developed a PPI assay based on the activity complementation of two mutant enzymes, FlimPIA (Firefly luminescent intermediate Protein-protein Interaction Assay) 1) 2).
1.An interaction assay based on firefly luciferase reaction mechanism
Probably, everyone has been once fascinated by the fantastic light emitted by fireflies. We have been interested in the light-emitting enzyme of firefly, luciferase (Fluc) and investigated its reaction mechanism 3). Recently, Fluc was reported to perform two-step enzyme reactions while changing its structure 4) 5). With its large N-terminal (N-) domain and smaller C-terminal (C-) domain linked via a flexible hinge, adenylation of luciferin (LH 2) is performed in a conformation to produce reaction intermediate luciferyl adenylate (LH 2-AMP). However, the oxidative luminescent reaction to produce oxyluciferin (OxL) from LH 2-AMP is performed in another conformation where C-domain is rotated by about 140° (Fig. 1A).
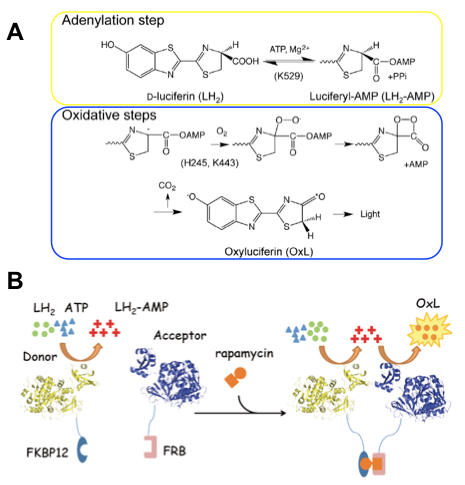 Figure 1. Scheme of firefly luciferase reaction and principle of FlimPIA.
Figure 1. Scheme of firefly luciferase reaction and principle of FlimPIA.
(A) Chemical reactions catalyzed by luciferase, which can be divided into two half reactions: adenylation and oxidative luminescent reactions. Catalytic residues are shown in the parentheses.
(B) Principle of FlimPIA. Fluc mutant whose oxidative luminescent reaction is slow (donor) and another mutant whose adenylation reaction is slow (acceptor) are used. When the donor and acceptor come close at the time of interaction, luminescence activity is enhanced by the relay of reaction intermediate LH 2-AMP.
This finding is primarily based on the evidence that the catalytically important C-domain residue in each half reaction is different. An important C-domain residue in adenylation is K529, while that in oxidative luminescent reaction is K443 locating at the other side of the domain, which strongly suggests C-domain rotation upon the reaction to proceed 6). Also we had found that oxidative luminescent reaction is selectively suppressed by mutating the active site residue H245 in N-domain to Asp 3). Based on these evidences, we reasoned that, by combining these mutations, one can make a PPI assay using two mutant enzymes, whose one of the half reactions is normal but the other half is abolished. When the two mutants come closer, the efficiency to transfer LH 2-AMP across them will be increased, thus that of overall reaction will be also increased (Fig. 1B). Consequently, as the "donor" of reactive intermediate, the Fluc mutant H245D / K443A was made, and as the "acceptor", a mutant K529Q was employed. To verify the system, as an interacting protein pair involved in inflammation, FK506 binding protein-12 (FKBP12) and FKBP12-rapamycin associated protein (FRB) were chosen and fused with the donor and acceptor enzymes, respectively, to make probes. These proteins are known to interact via an antibiotic rapamycin, an immunosuppressive drug also known for its anti-cancer activity. After purified probes were mixed and used for PPI assay in vitro, by adding rapamycin to the mixture, significant dose-dependent elevation in luminescent intensity was observed within 1 second after adding substrates LH 2 and ATP (Figure 2A). Also, by transfecting expression vectors to mammalian cells, similar rapamycin-dependent luminescence increase was detected (Figure 2B). Moreover, in the same manner we made the probes for p53-Mdm2 interaction, whose abnormality is observed in many cancer cells. The addition of Nutlin-3, a known inhibitor of the interaction, significantly decreased the light emission (Fig. 2C). The result also suggests a possibility that FlimPIA can be used for PPI inhibitor screening.
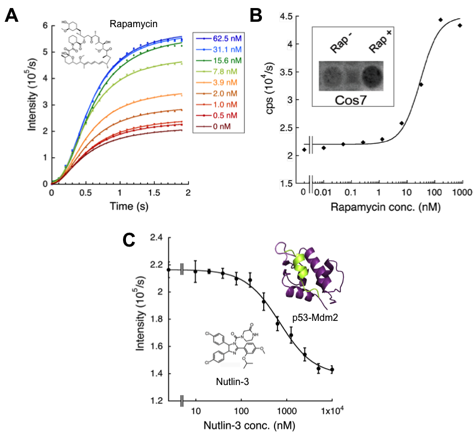 Figure 2. PPI detection by FlimPIA.
Figure 2. PPI detection by FlimPIA.
(A) Rapamycin concentration dependent FKBP12-FRB interaction detection in vitro. (Probe: 50 nM)
(B) Graph: interaction detection in the 293T cells. Frame within: Luminescent image of Cos7 cells (using LAS-4000mini).
(C) Inhibition of p53-Mdm2 interaction by Nutlin-3 (Probe: 50 nM).
2.Merits of FlimPIA
So what is the advantage of FlimPIA over the conventional methods? We first tried the in vitro detection of Protein-fragment Complementation Assay (PCA) as a comparison. As a result, it showed lower performance than FlimPIA in terms of signal strength and stability 2) 8). Then we tried a comparison with FRET between fluorescent protein (FP), which is often used to assay PPI inside and outside of the cells. As an interaction assay, how large protein pairs can be detected is an important parameter related to the versatility of the system. In other words, this is related to the detectable distance between the probes to generate sufficient signal. Detectable distance in FRET is dependent on the Förster distance R 0, and the signal is known to decrease within short distance in inverse proportion to the sixth power of the distance between the probes. Previously, if a Fibronectin Type III domain 7-8 (Fn7-8) having a rigid structure of 7 nm length is inserted between CFP and YFP, FRET signal is reported to become undetectable 9). Therefore, we made a similar insertion of Fn7-8 between one of the probe and FKBP12 in three FKBP12-FRB interaction assays, namely, FRET, PCA, and FlimPIA. As a result, in FRET and PCA, PPI detection was almost impossible. However, in the same condition FlimPIA did detect the interaction. The result clearly shows that FlimPIA has a superiority for detecting over longer distance (Fig. 3) 2).
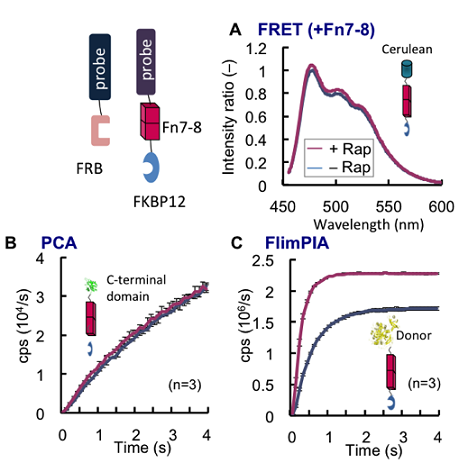 Figure 3. Comparison of the detectable probe distance A rigid linker of 7 nm length (Fn7-8) was inserted between FKBP12 and a probe. As a result, while FRET (A) and PCA (B) could not detect FKBP12-FRB interaction, FlimPIA (C) could do it under the same condition.
Figure 3. Comparison of the detectable probe distance A rigid linker of 7 nm length (Fn7-8) was inserted between FKBP12 and a probe. As a result, while FRET (A) and PCA (B) could not detect FKBP12-FRB interaction, FlimPIA (C) could do it under the same condition.
3.For better FlimPIA response
At its initial stage of development, there was a problem in FlimPIA that the background emission of the acceptor enzyme without interaction resulted in lower signal increase at the time of interaction (i.e. low S / B ratio). As mentioned above, Fluc performs the two half-reactions while changing its structure. So we reasoned that if we fix the conformation of the acceptor enzyme to the oxidative luminescent reaction structure, it would be possible to reduce the residual adenylation activity. Therefore, pairs of adjacent residues in the oxidative structure, one each in the N- and C-domains of the acceptor, were replaced by cystine, and these were cross-linked by thiol-specific crosslinker or simply by disulfide bonds, to fix the enzyme to oxidative reaction structure. As a result, we could successfully improve the S/B ratio 2). More recently, we also succeeded to further improve the response by the mutagenesis and reaction condition optimization. In future, we hope more interactions to be more robustly and reliably detected.
4.Summary
Here we showed that by dividing an enzyme in terms of chemical reactions rather than by physical means, it becomes a useful reagent for PPI detection. Since it is compatible with PPI involving large proteins in vitro, in near future it will be applied to quicker clinical diagnosis. In addition, since similar approach will be possible for other ANL superfamily enzymes, a novel application as a new enzyme activity control method in synthetic biology is also expected.
References
1) Ohmuro-Matsuyama Y et al., Anal. Chem., 85, 7935-7940 (2013)
2) Ohmuro-Matsuyama Y et al., ibid., 86, 2013-2018 (2014)
3) Ayabe K et al., FEBS Lett., 579, 4389-4394 (2006)
4) Nakatsu, T., et al., Nature, 440, 372-376 (2006)
5) Branchini BR et al., J. Am. Chem. Soc., 133, 11088-11091 (2011)
6) Branchini BR et al., Biochemistry, 51, 6493-6495 (2012)
7) Branchini BR et al., ibid., 44, 1385-1393 (2005)
8) Ohmuro-Matsuyama Y et al., BMC Biotechnol. 13, 31 (2013)
9) Ohashi T et al., Protein Sci., 16, 1429-1438 (2007)
